Fabry-Krankheit
Die Fabry-Krankheit ist eine lysosomale Speicherkrankheit, die X-chromosomal rezessiv vererbt wird und durch eine Mutation im GLA-Gen verursacht wird. Aufgrund dieser Mutation besteht ein Mangel an dem Enzym Alpha-Galaktosidase A, das für den Abbau fetthaltiger Substanzen verantwortlich ist. Dies führt zur Ansammlung von Globotriaosylceramid in verschiedenen Organen.
Die Fabry-Krankheit gehört zur Gruppe der ultra-seltenen Mucopolysaccharidosen. Die Erkrankung kann zu schweren Anfällen mit quälenden, brennenden Schmerzen in den distalen Extremitäten führen, die in andere Körperbereiche ausstrahlen.
Typische Symptome sind außerdem Störungen der Thermoregulation und des Schwitzens, selbst in Stresssituationen. Betroffene versterben häufig vor dem 50. Lebensjahr an vorzeitigem Herz- oder Hirnschaden. Aufgrund der unspezifischen Symptomatik kann es Jahre dauern, bis eine korrekte Diagnose gestellt wird.
Ursachen der Fabry-Krankheit
Die Fabry-Krankheit ist eine seltene lysosomale Speicherkrankheit. Sie wird durch eine Mutation im GLA-Gen verursacht, wodurch die Aktivität des Enzyms Alpha-Galaktosidase A entweder vollständig fehlt oder stark reduziert ist.
Infolge dessen kommt es zur Anreicherung von Globotriaosylceramid (GL-3) in den Zellen verschiedener Gewebe und Organe, da der Enzymmangel dessen Abbau verhindert. Die Akkumulation von GL-3 beeinträchtigt die Organfunktionen und führt zu Komplikationen.
Diese seltene Erkrankung ist X-chromosomal vererbt und tritt daher häufiger bei Männern auf. Heterozygote Frauen können jedoch ebenfalls Symptome zeigen, deren Schweregrad individuell unterschiedlich ist. Bei Frauen manifestiert sich die Erkrankung in der Regel später als bei Männern.
Diagnose der Fabry-Krankheit
Zur Diagnosesicherung bei Männern wird die Aktivität der Alpha-Galaktosidase A in Plasma, Leukozyten oder Fibroblasten gemessen. Bei nachgewiesenem Enzymmangel erfolgt eine genetische Analyse.
Bei Frauen kann die Enzymaktivität im Normbereich liegen – hier ist eine genetische Untersuchung notwendig, um Trägerinnen des defekten Gens zu identifizieren.
Symptome der Fabry-Krankheit
Die Erkrankung tritt in zwei Erscheinungsformen auf:
- Klassischer Phänotyp – häufiger bei Männern, mit fehlender oder minimaler Enzymaktivität. Die Symptome beginnen im Kindes- oder Jugendalter.
- Nicht-klassischer Phänotyp – mit späterem Symptombeginn aufgrund einer milderen Enzymdefizienz.
Symptome im Jugendalter:
- Hitzetoleranzstörungen
- Gestörtes Schwitzen
- Hyperthermie
- Parästhesien (intensiv brennende Schmerzen in den Gliedmaßen)
- Bauchschmerzen, Verstopfung, Durchfall, Übelkeit und Erbrechen
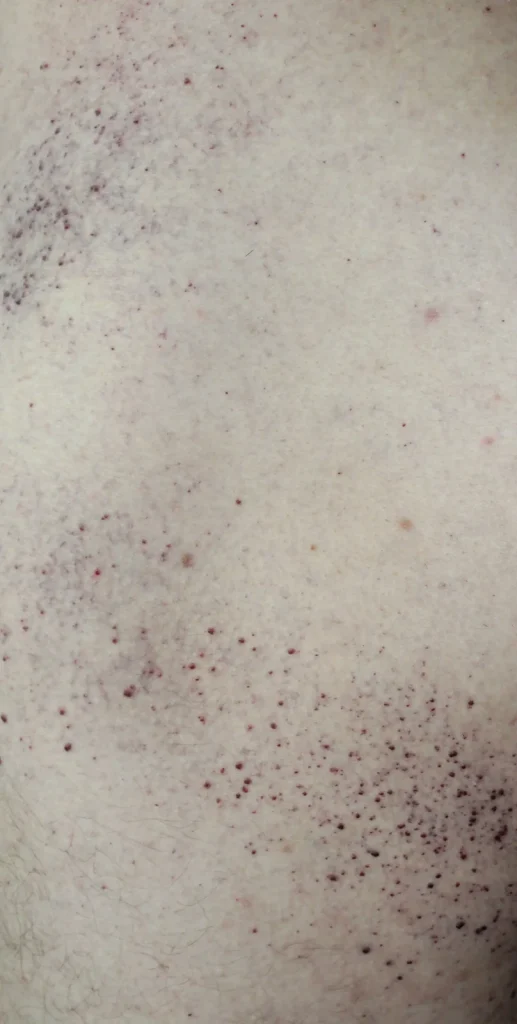
- Hautveränderungen an Oberschenkeln, Gesäß und Unterbauch (Angiokeratome – rotviolette Ausschläge)
- Hornhauttrübung, Katarakte, fortschreitender Hörverlust
Symptome im Erwachsenenalter:
- Strukturelle und funktionelle Herzveränderungen (Linksherzhypertrophie, Arrhythmien, Reizleitungsstörungen)
- Nierenschäden, zunächst erkennbar durch Proteinurie und Mikroalbuminurie
- Chronische Müdigkeit, Tinnitus, Schwindel
Verhaltensänderungen wie Angstzustände, soziale Isolation, Depression, Schuldgefühle.
Viele dieser Symptome treten bereits lange vor der Diagnose auf. Wie bei anderen seltenen Erkrankungen erleben auch Fabry-Patienten häufig lange diagnostische Verzögerungen, was sich negativ auf die körperliche und psychische Gesundheit auswirkt. Studien zeigen, dass die Lebensqualität von Fabry-Betroffenen deutlich geringer ist als in der Allgemeinbevölkerung.
Prognose der Fabry-Krankheit
Die häufigsten Todesursachen bei Fabry-Patienten sind Schlaganfall und Herzinfarkt. Männer mit Fabry-Krankheit haben eine um 15–20 Jahre verkürzte Lebenserwartung, Frauen leben im Schnitt 6–10 Jahre kürzer als die Allgemeinbevölkerung.
Behandlung der Fabry-Krankheit
Die Standardtherapie besteht in der Enzymersatztherapie (ERT) mit Agalsidase alfa oder beta. Aufgrund der multisystemischen Ausprägung der Krankheit sind ergänzende Therapien notwendig, um Komplikationen zu behandeln und den Gesundheitszustand zu stabilisieren.
ERT kann den Krankheitsverlauf verlangsamen und es den Patienten ermöglichen, familiäre, berufliche und soziale Aktivitäten beizubehalten. In Polen wird ERT seit 2019 im Rahmen eines medizinischen Programms erstattet.
Zusätzlich ist Migalastat zur Behandlung zugelassen – ein orales Medikament, im Gegensatz zur intravenösen ERT. Es ist jedoch nur für Patienten mit spezifischen, auf das Medikament ansprechenden Mutationen geeignet (ca. 35–50 % aller Betroffenen). Seit 2020 ist Migalastat in Polen für Patienten über 16 Jahren mit bestätigter Diagnose und geeigneter Mutation verfügbar.
Unterstützende Therapien bei Fabry:
- Schmerztherapie (pharmakologisch)
- Neuroprotektion (ACE-Hemmer und Angiotensin-Rezeptorblocker)
- Antiarrhythmika
- Herzschrittmacher oder implantierbarer Defibrillator (ICD) bei schweren Herzbeteiligungen
- Dialyse oder Nierentransplantation bei fortgeschrittener Nierenschädigung
Die Behandlung ist lebenslang. Eine frühzeitige Intervention erhöht die Chancen, schwere Komplikationen hinauszuzögern oder zu verhindern.
Häufigkeit der Fabry-Krankheit
Bis 2018 wurde die Fabry-Krankheit in Polen bei ca. 73 Personen diagnostiziert. Weltweit liegt die geschätzte Prävalenz zwischen 1:40.000 und 1:170.000. Aufgrund der komplexen Symptomatik und der schwierigen Differenzialdiagnose könnte die tatsächliche Zahl der Betroffenen jedoch höher sein.